БОЛЕЗНЬ ФАБРИ
Эта страница посвящена болезни Фабри —
одному из наиболее распространенных
заболеваний группы лизосомных болезней
накопления1-3.

также известная как болезнь Андерсона-Фабри — это Х-сцепленная лизосомная болезнь накопления (ЛБН). Впервые это заболевание описано в 1898 г.4-6 Причиной развития болезни Фабри является мутация гена GLA, кодирующего фермент альфа-галактозидазу А (α-Gal A). Дефицит активности фермента α-Gal A приводит к накоплению в клетках гликосфинголипидов — глоботриаозилцерамида (Gb3) и глоботриаозилсфингозина (лизо-Gb3). Это вызывает поражение различных органов и тканей3, 7-11.
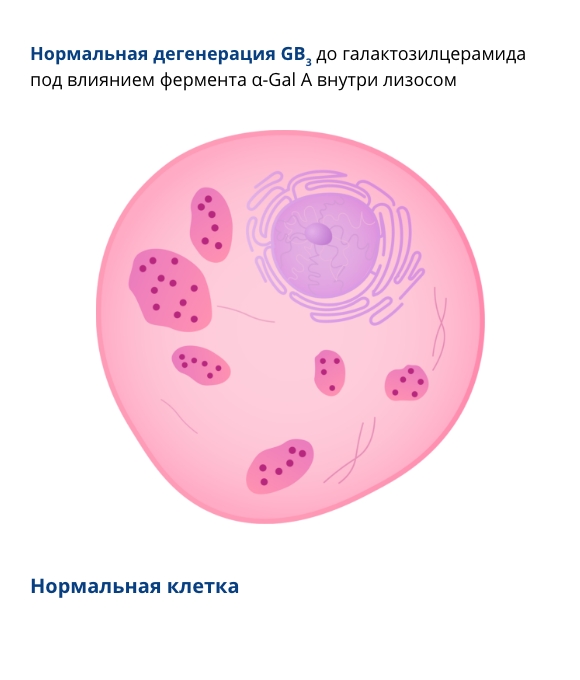
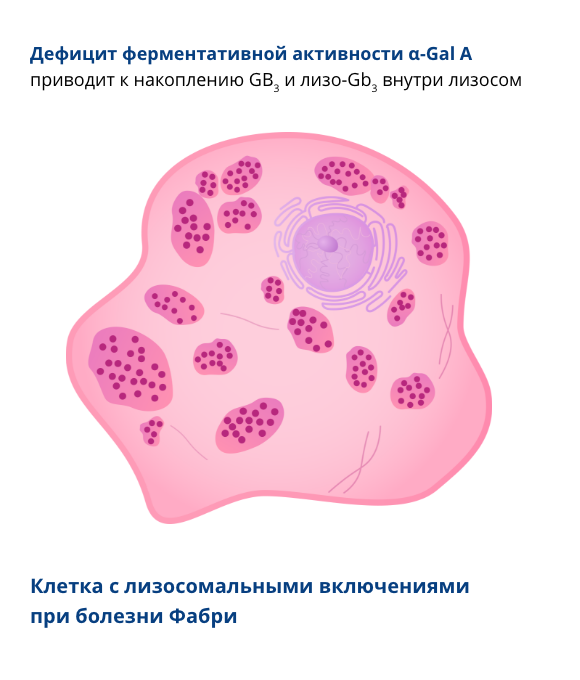
Рисунок 1. Мутация гена GLA, кодирующего фермент α-Gal A, приводит к накоплению гликосфинголипидов (Gb3 и лизо-Gb3) в различных органах пациентов с болезнью Фабри.
Источник: изображение взято и адаптировано из открытого интернет-ресурсаfabry-institute.com



наблюдается во всех этнических, расовых и демографических группах и поражает примерно 1 из 40 000 мужчин и 1 из 20 000 женщин11,16.

Систематический обзор скрининговых исследований новорожденных показал, что распространенность мутаций GLA у новорожденных составляла 0,04%, что соответствует заболеваемости болезнью Фабри 1 на 2597 живорожденных. Таким образом, данные программ скрининга новорожденных свидетельствуют о том, что заболеваемость болезнью Фабри, как правило, недооценивается и может быть выше17.
Х-сцепленная лизосомная болезнь накопления. Х-сцепленные заболевания с рецессивным типом наследования обычно проявляются у мужчин, а лица с пораженным аллелем на Х-хромосоме являются гемизиготными по заболеванию и не могут передать заболевание своим потомкам мужского пола3.
Однако все потомки женского пола будут облигатными гетерозиготами по заболеванию. Гетерозиготная женщина, не пораженная болезнью, с 50-процентной вероятностью может передать заболевание потомству мужского пола, тогда как её потомство женского пола с такой же вероятностью станет облигатными гетерозиготами по заболеванию18.
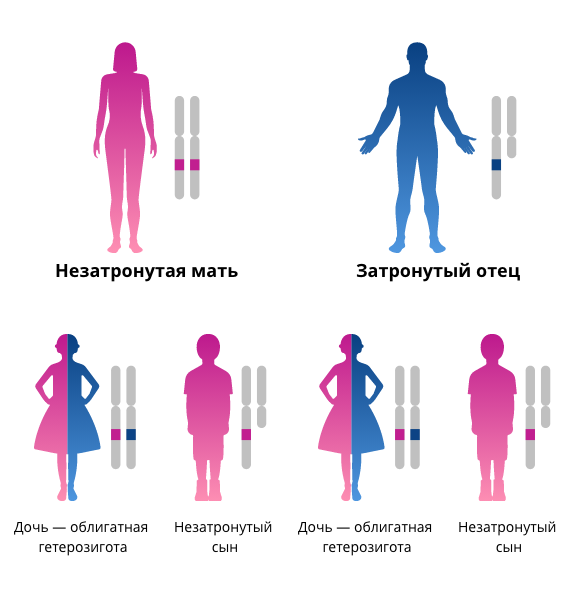
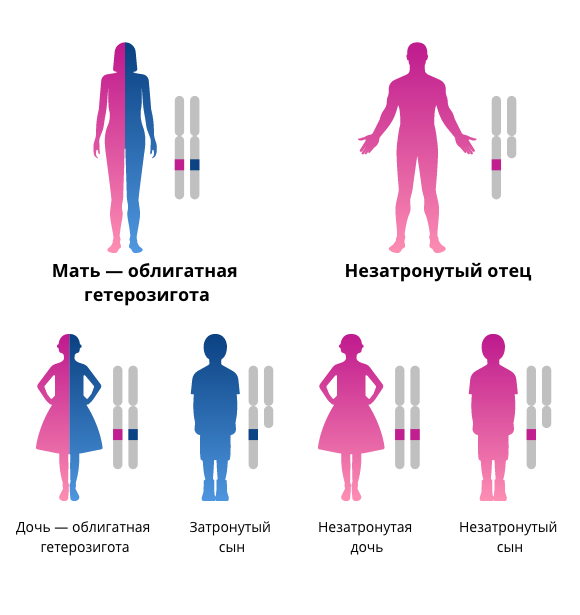
Рисунок 2. Генетическое наследование болезни Фабри.
Источник: изображение взято и адаптировано из открытого интернет-ресурсаfabry-institute.com
Из-за X-связанного наследования болезни Фабри женщины исторически считались «бессимптомными гетерозиготами». В настоящее время признано, что у женщин с болезнью Фабри может проявляться разнообразный спектр проявлений болезни, которые могут быть такими же тяжелыми, как и у пациентов мужского пола19-22. На сегодняшний день идентифицировано более 1000 вариантов мутаций гена GLA, приводящих к дефициту активности α-Gal A9,10,23,24. Большинство вариантов мутаций GLA являются частными, встречающимися в одной или нескольких семьях. Имеют место некоторые корреляции между генотипом и фенотипом болезни Фабри. Например, миссенс-мутация N215S (c.644A>G) постоянно наблюдается у пациентов с преимущественно сердечными проявлениями болезни. И наоборот, поражение почек и сосудов головного мозга у пациентов с этим вариантом мутации встречается редко25,26. Считается, что у женщин с Х-сцепленными заболеваниями, такими как болезнь Фабри, тяжесть проявлений заболевания зависит от уровня инактивации Х-хромосомы27.
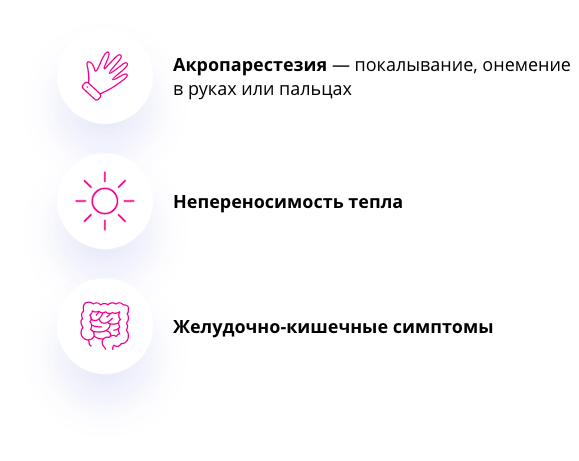
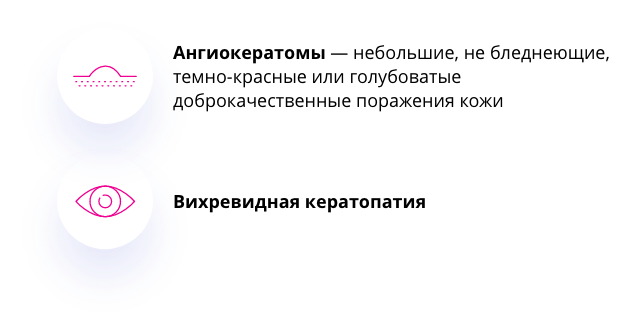

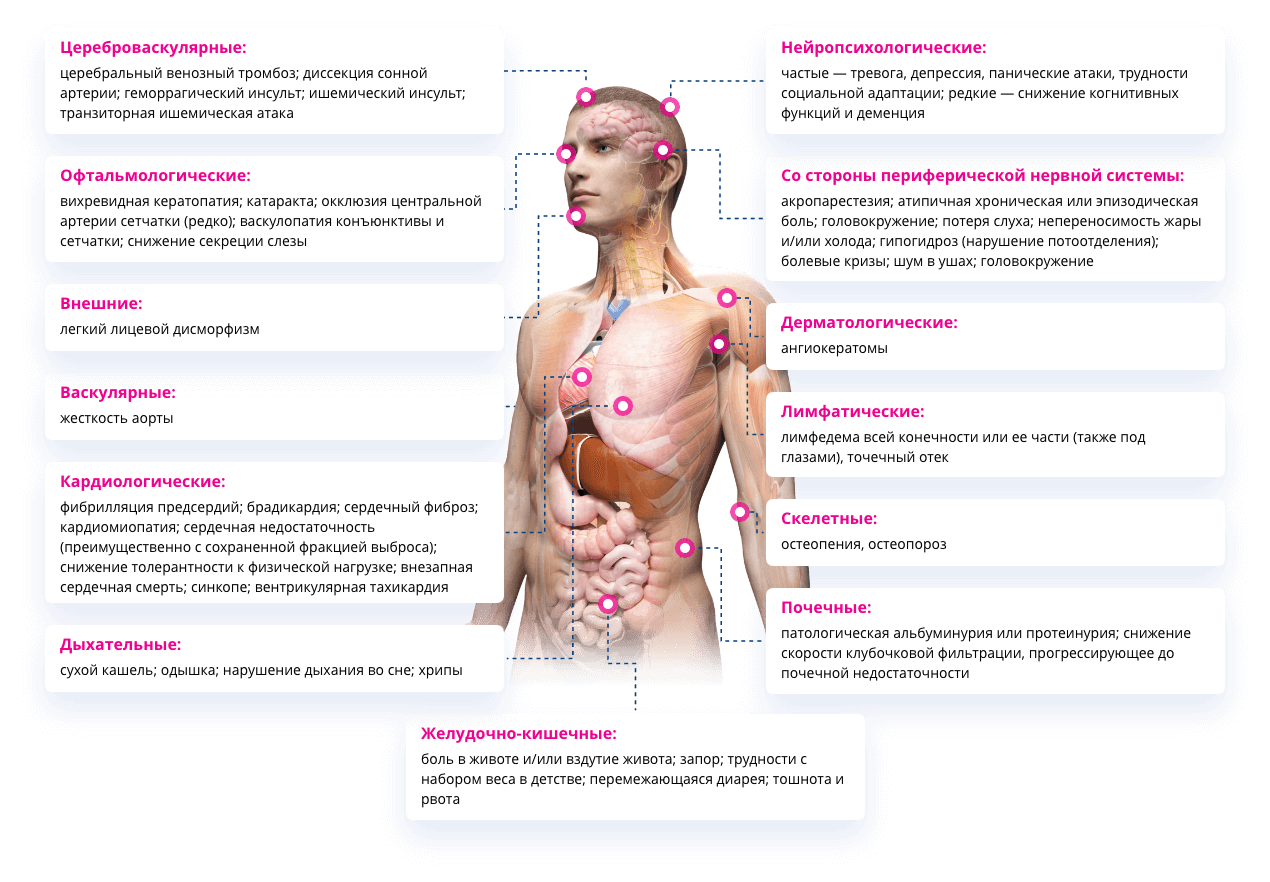
Рисунок 3. Клинические проявления классической болезни Фабри.
Источник: изображение взято и адаптировано из открытого интернет-ресурсаfabry-institute.com
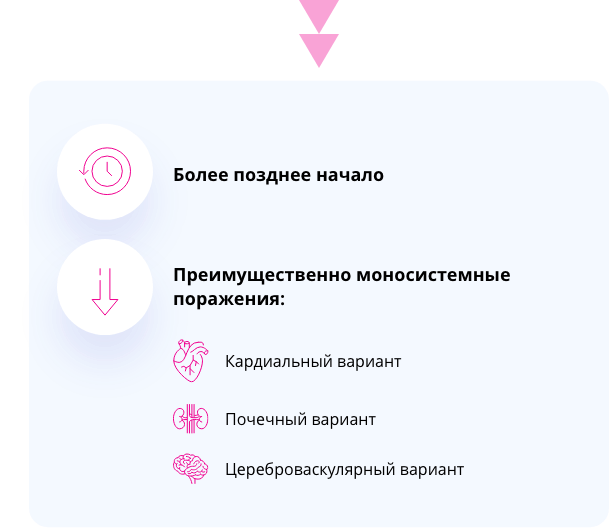
характеризируется более медленным прогрессированием и вариабельностью проявлений по сравнению с классической болезнью Фабри. Поражение может быть менее выраженным, а проявления болезни могут быть ограничены только одним органом31. Типичные сердечные и почечные симптомы могут проявиться только в более позднем возрасте (на четвертом-восьмом десятилетии жизни), отражая отсроченное начало заболевания. Болезнь может быть выявлена случайно14, 25, 26, 32, 33.



Источник: изображения взяты и адаптированы из открытого интернет-ресурсаwww.shutterstock.com
до диагностики заболевания часто выставляются неверные диагнозы. Согласно опубликованном в 2004 г. исследовании 366 пациентов с болезнью Фабри, включенных в реестр Fabry Outcome Survey (FOS)*,7, 39:
пациентов прежде были установлены неверные диагнозы
составляет средняя задержка от появления симптомов до установки правильного диагноза болезни Фабри у мужчин
составляет средняя задержка от появления симптомов до установки правильного диагноза болезни Фабри у женщин
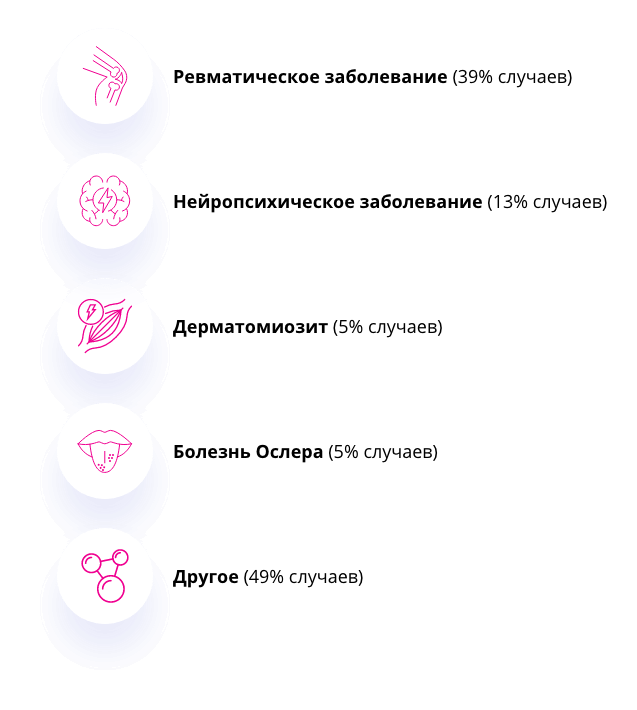
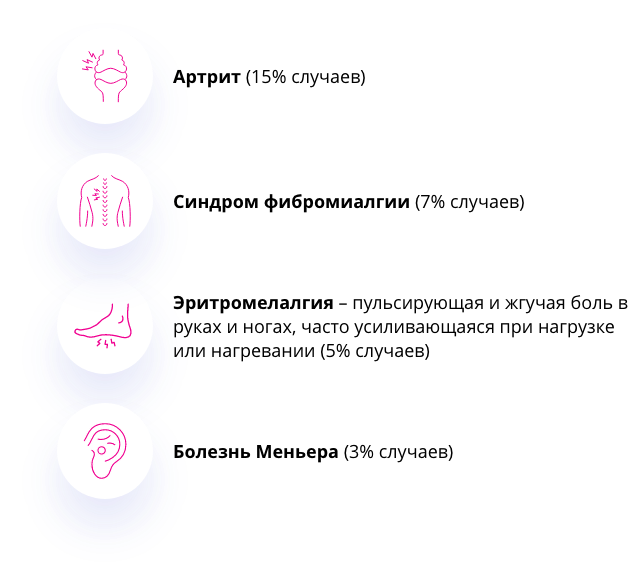
В некоторых случаях болезнь Фабри также может быть неправильно диагностирована как рассеянный склероз, потому что у пациентов в обоих случаях может иметь место болевой синдром и поражения белого вещества, определяемые при магнитно-резонансной томографии40.
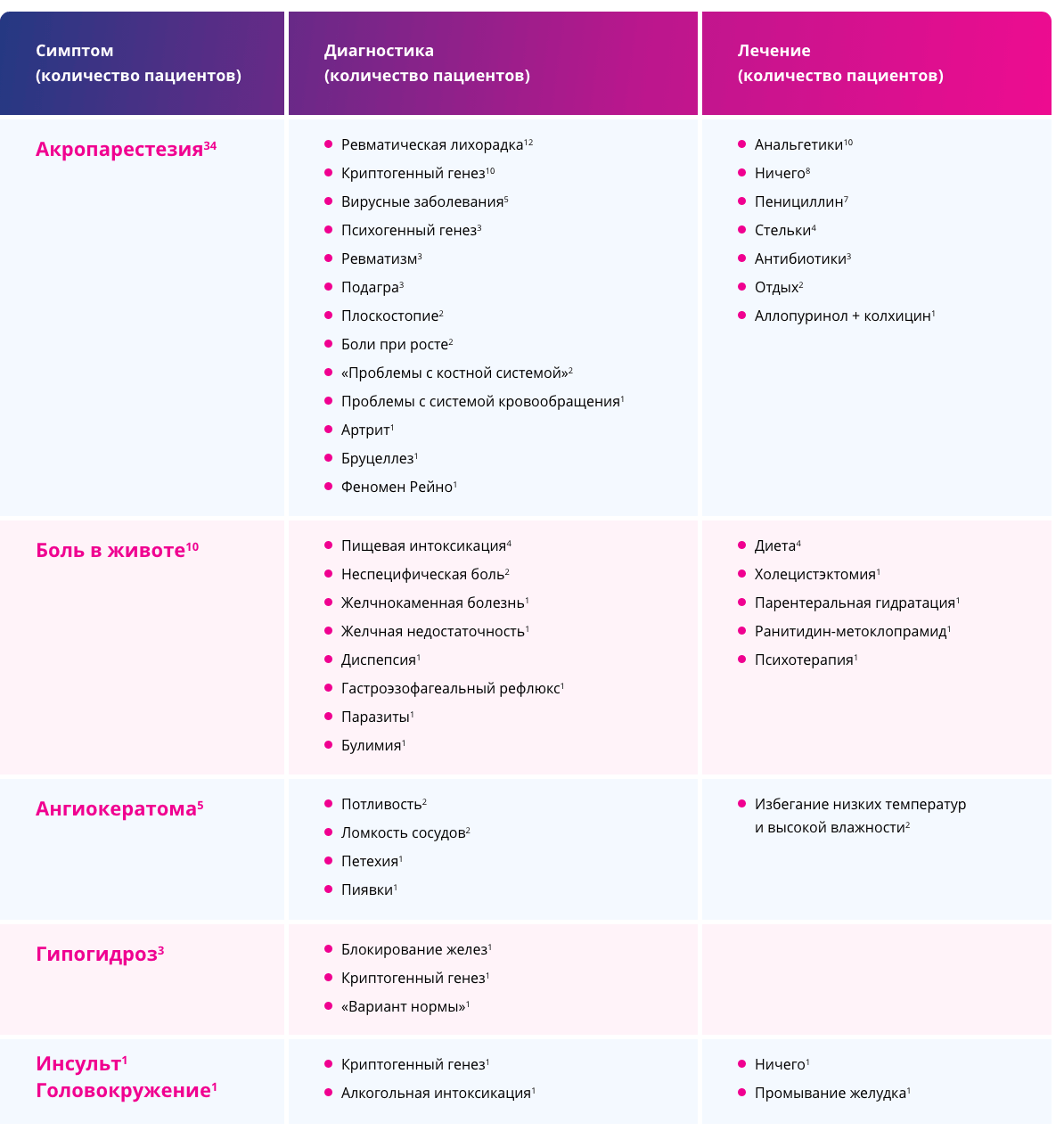
Рисунок 4. Ошибочный диагноз болезни Фабри: симптомы, диагностические ошибки и последующее лечение.
Источник: изображение взято и адаптировано из открытого интернет-ресурсаfabry-institute.com
Результаты исследований показывают, что ошибочный диагноз болезни Фабри является распространенным явлением. Болезнь Фабри должна быть включена в дифференциальную диагностику пациентов с акропарестезией, гипогидрозом и рецидивирующей болью в животе в детском или подростковом возрасте.
*FOS — это реестр заболеваний, спонсируемый компанией Takeda, который собирает информацию о пациентах с болезнью Фабри по всему миру. Информация о симптомах, прогрессировании заболевания и лечении собраны с тех пор, как первые пациенты были включены в FOS в 2001 году. В настоящее время зарегистрировано более 4000 пациентов. Цель FOS — улучшить понимание болезни Фабри, что поможет улучшить оказание медицинской помощи данной группе пациентов42.
представляет собой сложное полисистемное заболевание с широким спектром клинических проявлений, включая:

Дерматологические

Офтальмологические

Сердечно-сосудистые

Почечные

Неврологические
Следовательно, болезнь Фабри следует учитывать при дифференциальной диагностике многих системных заболеваний. Тем не менее, диагностика болезни Фабри может быть затруднена из-за разнообразия клинических симптомов43.




Источник: изображение взято и адаптировано из открытого интернет-ресурсаwww.shutterstock.com
Согласно Канадскому руководству по лечению болезни Фабри 2018 г., никакие клинические проявления болезни Фабри не являются диагностическими сами по себе. Некоторые признаки заболевания (например, нефропатия, гипертрофическая кардиомиопатия и инсульт) неспецифичны и требуют различных дифференциальных диагнозов. Некоторые проявления болезни Фабри имеют более ограниченный дифференциальный диагноз (например, вихревидная кератопатия, ангиокератомы), однако эти симптомы все еще могут присутствовать при других состояниях47.
Предварительный диагноз болезни Фабри может быть установлен на основании клинических проявлений у конкретного пациента, поскольку спектр возможных дифференциальных диагнозов широк. Клинические данные сами по себе не могут быть использованы для подтверждения диагноза болезни Фабри. В случае сомнений клиницисты должны включить болезнь Фабри в число возможных диагнозов48.
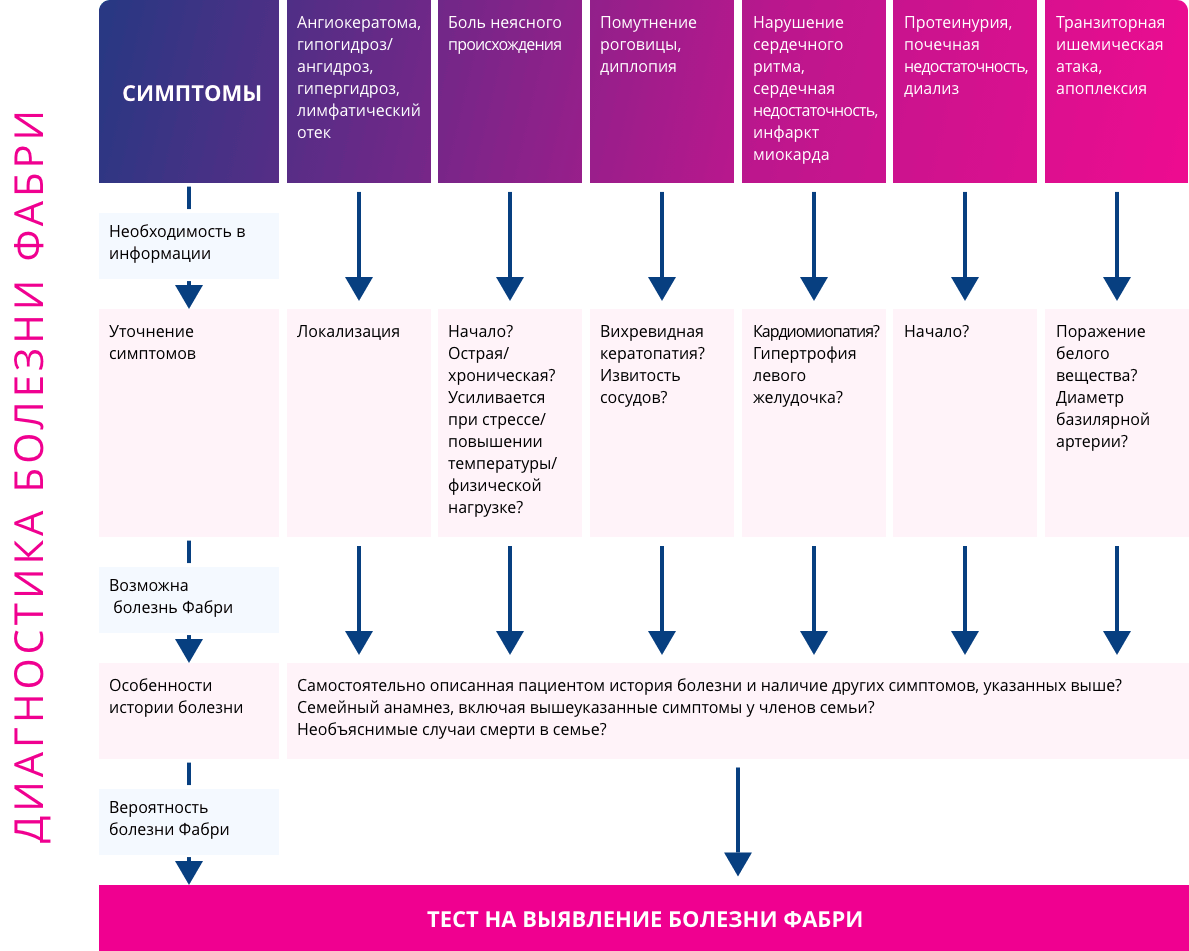
Рисунок 5. Блок-схема диагностики болезни Фабри: от неспецифических симптомов до постановки диагноза.
Источник: открытый интернет-ресурсfabry-institute.com
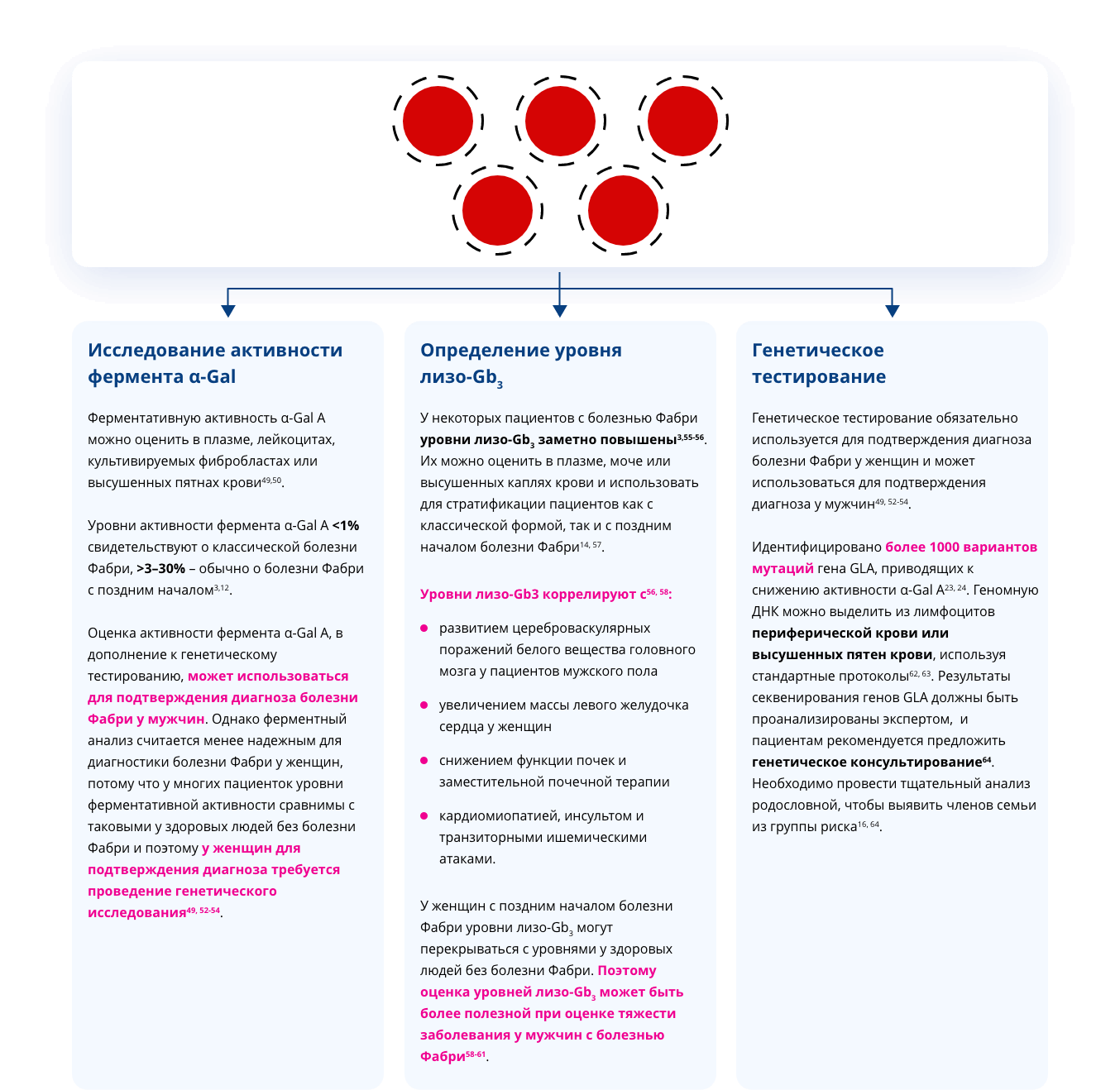
01
У пациента с подозрением на болезнь Фабри берут образец СКК* и отправляют его в специализированную лабораторию для исследования
02
По СКК выполняется генетическое исследование — наличие мутации, активность фермента, наличие субстрата Gb3**
03
Лаборатория отсылает результаты исследования лечащему врачу
*СКК — сухая капля крови
**Gb3 — глоботриазилцерамид

После установления диагноза болезни Фабри рекомендуется провести семейный скрининг, чтобы обнаружить всех членов семьи, имеющих риск развития заболевания. Одним из преимуществ семейного скрининга является возможность выявление членов семьи на ранних стадиях заболевания. Следовательно, семейный скрининг может сократить время до постановки диагноза с момента появления симптомов, исключить ошибочные диагнозы и, при необходимости, обеспечить раннее лечение16, 64-66.

Семейный скрининг на болезнь Фабри должен проводиться медицинскими работниками, имеющими опыт генетического консультирования. Рекомендуется провести полный анализ родословной, включающий ≥3 поколений67.

Члены семьи, у которых впоследствии диагностируется болезнь Фабри, также должны получать поддержку и консультации65, 67.
является предотвращение необратимого повреждения тканей и органной недостаточности. Рекомендуется, чтобы оценка лечения и последующие визиты проводились под наблюдением врача, имеющего опыт лечения болезни Фабри и, если это возможно, при участии многопрофильной команды, включающей кардиолога, генетика, нефролога, невролога, медсестру и психолога14.


недостаточная активность фермента α-Gal A приводит к накоплению гликосфинголипидов Gb3 и лизо-Gb3 почти во всех типах клеток и различных органах8, 9, 10. Заместительная ферментная терапия заключается в введении рекомбинантного фермента α-Gal A, что помогает устранить основной дефицит фермента и восстановить расщепление накопленного Gb3 у пациентов с болезнью Фабри80.
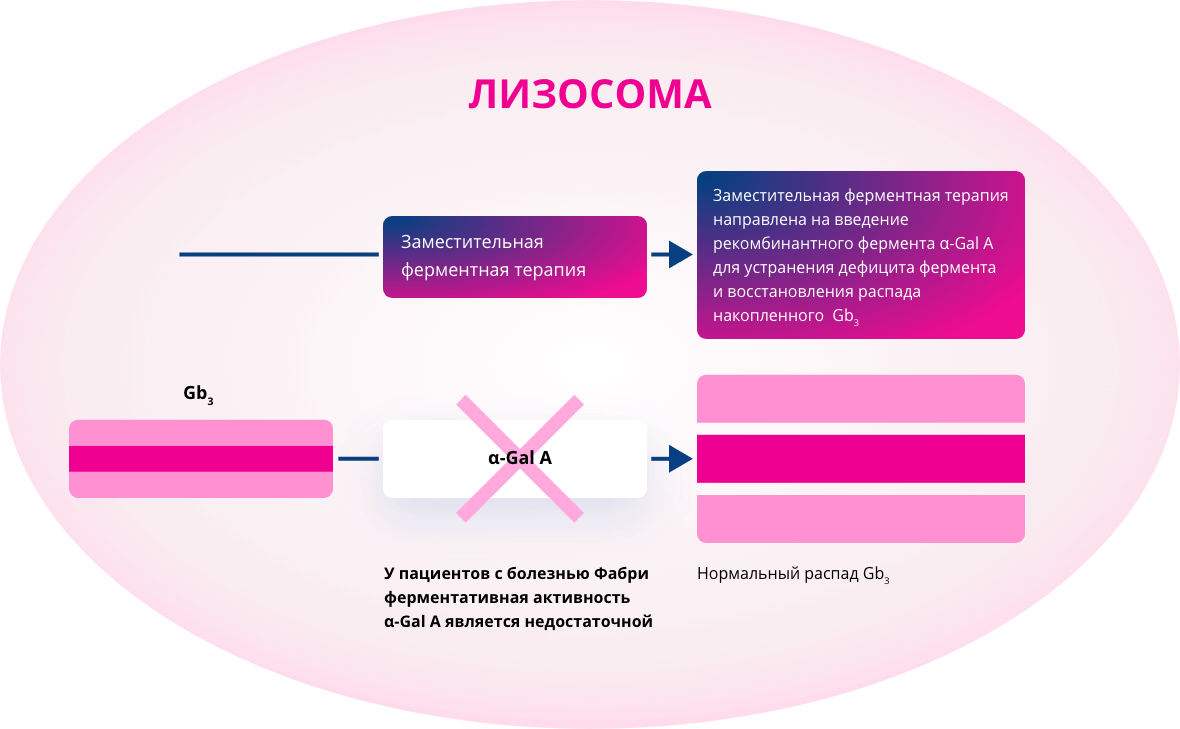
α-Gal A — альфа-галактозидаза А; Gb3 — глоботриаозилцерамид.
Рисунок 7. Заместительная ферментная терапия восстанавливает распад Gb3 у пациентов с болезнью Фабри.
Источник: изображение взято и адаптировано из открытого интернет-ресурсаfabry-institute.com
внутривенная ферментозаместительная терапия для пациентов с болезнью Фабри. Агалсидаза альфа показана для длительного лечения пациентов с болезнью Фабри. Агалсидаза альфа замещает лизомосальный фермент α-галактозидазу А и приводит к уменьшению накопления Gb3 в клетках, включая эндотелиальные и паренхимальные клетки. Это приводит к облегчению симптомов и улучшению качества жизни у пациентов с болезнью Фабри69.
1. У пациентов с болезнью Фабри недостаточная активность фермента a-Gal A приводит к накоплению Gb3 и лизо-Gb3 в лизосомах
2. Агалсидаза альфа катализирует гидролиз Gb3, отщепляя от молекулы терминальный фрагмент галактозы
3. Клетка, получающая заместительную ферментную терапию агалсидазой альфа
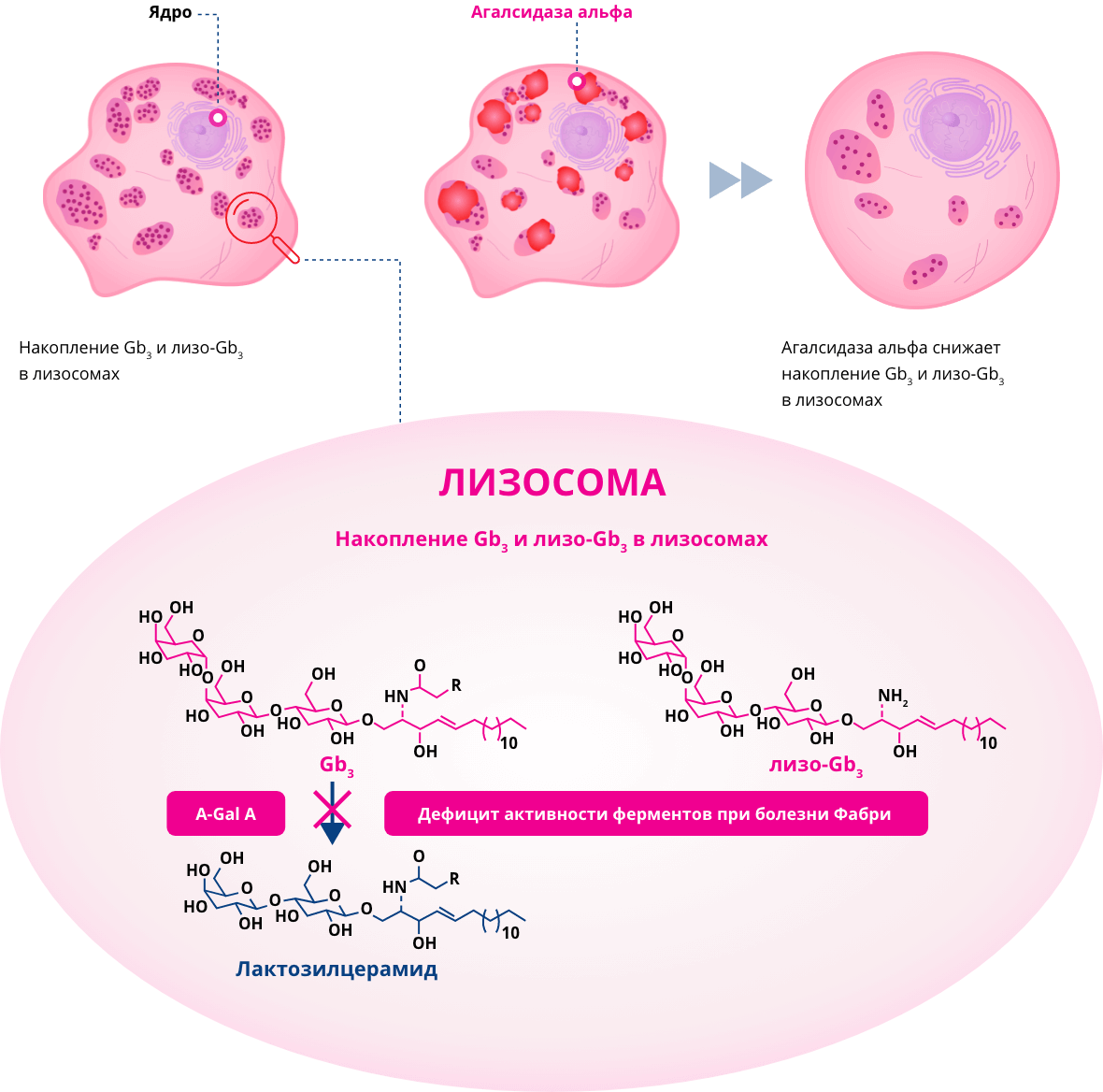
α-Gal A — альфа-галактозидаза А; Gb3 — глоботриаозилцерамид; лизо-Gb3 — глоботриаозилсфингозин.
Рисунок 8. Механизм действия агалсидазы альфа.
Источник: изображение взято и адаптировано из открытого интернет-ресурсаfabry-institute.com
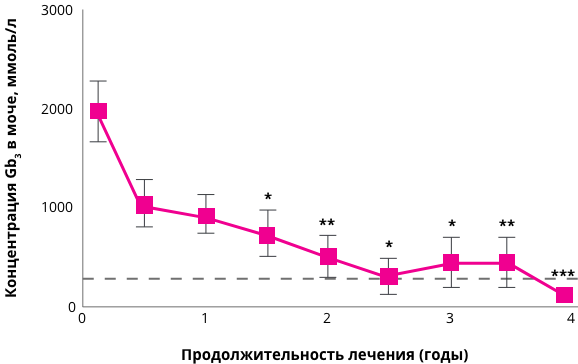
Пунктирная линия — среднее значение показателя среди здорового населения
Применение агалсидазы альфа ассоциируется со значительным снижением уровня GL-3 в моче на протяжении всего периода лечения и достижения референтных значений81
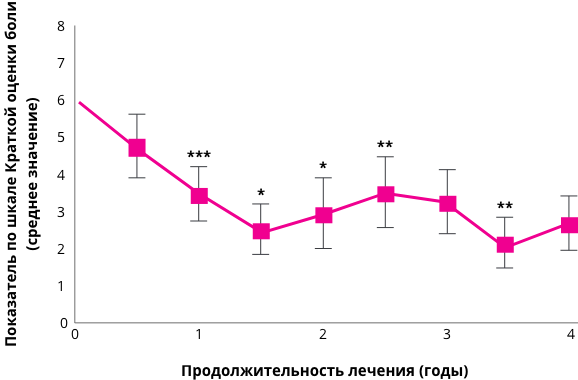
Применение агалсидазы альфа ассоциируется со снижением нейропатической боли на 53% уже после первого года лечения81
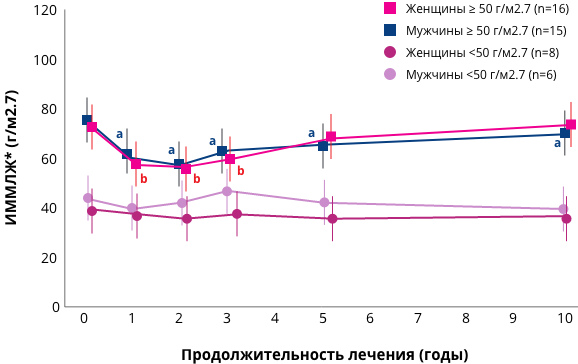
*ИММЛЖ — индекс массы миокарда левого желудочка
Применение агалсидазы альфа ассоциируется с долгосрочным сохранением структуры сердца82
Изображение адаптировано из публикации Kampmann С и др.1
a Статистически значимое (Р <0.05) изменение от исходного уровня среди мужчин с ИММЛЖ ≥50 г/м2.7 до лечения.
b Статистически значимое (Р <0.05) изменение от исходного уровня среди женщин с ИММЛЖ ≥50 г/м2.7 до лечения.
Данные из одного центра. Пациенты мужского и женского пола были разделены по ИММЛЖ < 50 или ≥ 50 г/м2.7 до лечения. Точки данных — это средства с SD.
Применение агалсидазы альфа ассоциируется с улучшением или стабилизацией симптомов сердечной недостаточности и стенокардии в течение 10 лет82
После ~10 лет применения препарата агалсидазы альфа показатели классификации NYHA и CCS улучшились или остались стабильными у

В начале лечения симптомы сердечной недостаточности (NYHA класс ≥ II; указывает на легкую отдышку и/или стенокардию с обычной физической активностью2) наблюдались у 32% пациентов, а симптомы стенокардии (CCS класс ≥ 2; указывает на стенокардию с интенсивной или обычной физической активностью3) наблюдались у 25% пациентов. Рассчитано по классу улучшения или стабилизации NYHA в популяции (n=42), для которой имелись 10-летние данные: (22/42 × 100 = 52,4%) + (19/42 × 100 = 45,2%) = 97,6%, а также по классу улучшения или стабилизации CCS: (15/42 × 100 = 35,7%) + (26/42 × 100 = 61,9%) = 97,6%.
Применение агалсидазы альфа ассоциируется с долгосрочной стабилизацией или улучшением показателей сердечной функции82, 83
Применение агалсидазы альфа способствует долгострочному сохранению функции почек — за 10 лет показатели рСКФ были относительно стабильны у женщин и продемонстрировали небольшой спад у мужчин84.

Применение агалсидазы альфа ассоциируется со снижением риска сердечных, почечных или цереброваскулярных нарушений85
Вероятность возникновения заболевание (сердечно-сосудистого, цереброваскулярного, почечного или смерти)* (%) через 24 месяца после начала ферментозаместительной терапии
Для сравнения с пациентами из исследования Баникахеми М, и др.2, не проходившими лечение, конечная точка включала инфаркт миокарда, любое серьезное нарушение со стороны сердца, указывающее на ишемическую болезнь сердца, сердечную недостаточность, клапанный порок или аритмию; любое нарушение, приводящее к чрескожной транслюминальной коронарной ангиопластике или аортокоронарному шунтированию; любое серьезное событие сердечной недостаточности; серьезные нарушения, требующие проведения операции на клапанах; серьезные неблагоприятные события, указывающие на необходимость пересадки почки, проведения диализа или требующие проведения регулярного диализа; повышение содержания креатинина в сыворотке крови на 33% от исходного уровня (два последовательных значения); острое нарушение мозгового кровообращения, инсульт или преходящее ишемическое нарушение мозгового кровообращения, или смерть.
Предполагаемая средняя выживаемость составила 77,5 лет для пациентов мужского пола, получавших агалсидазу альфа, и 60 лет для пациентов контрольной группы без лечения в анамнезе85
Своевременное начало приема агалсидазы альфа ассоциируется с лучшими результатами в сравнении с отсроченным лечением87*
Своевременное начало приема агалсидазы альфа после постановки диагноза было связано со значительным снижением риска сердечно-сосудистых нарушений (HR, 0.768; 95% CI, 0.662-0.892), нарушений со стороны почек (HR, 0.779; 95% CI, 0.658-0.922) и комбинированных нарушений (HR, 0.836; 95% CI, 0.731-0.958) по сравнению с поздним началом лечения87*
* Ретроспективный анализ данных, полученных из исследования FOS. В группе оперативного начала лечения (n=934) прием агалсидазы альфа начинался до ≤ 2лет после постановки диагноза, а в группе позднего начала лечения (n=1002) прием агалсидазы альфа начинался через >2 года после постановки диагноза. Конечные точки в отношении сердечно-сосудистой системы включали инфаркта миокарда, гипертрофию левого желудочка и сердечную недостаточность. Конечные точки в отношении почек включали диализ (перитонеальный диализ, гемодиализ или неуточненный диализ), трансплантацию, почечную недостаточность и протеинурию (регистрируемую в FOS как признак/симптом). Комбинированные конечные точки включали нарушения со стороны почек и сердечно-сосудистой системы, и смерть.
Для контроля клинических проявлений, связанных с болезнью Фабри, симптоматическое лечение и поддерживающая терапия могут проводиться в дополнение к терапии, специфичной для болезни Фабри14.

